The Food and Drug Administration (FDA) is responsible for approving and regulating all drugs in the US. Aside from its domestic role, the FDA is also globally important, writes Robin Forrest. Drawing attention to mechanisms of global drug development, market dynamics, and recent trends in drug approvals, he argues that the influence of the FDA outside of the US cannot be overstated.
The Food and Drug Administration (FDA) is responsible for approving and regulating all drugs in the US. Aside from its domestic role, the FDA is also globally important, writes Robin Forrest. Drawing attention to mechanisms of global drug development, market dynamics, and recent trends in drug approvals, he argues that the influence of the FDA outside of the US cannot be overstated.
Amid the COVID-19 pandemic, the last few years of US politics and pharmaceutical research and development has been turbulent to say the least. But in this landscape, the US Food and Drug Administration (FDA) has remained steadfast on approving new drugs quickly for US patients. Looking at media coverage of the most high-profile cases, some approvals have been met with controversy, whilst others have been broadly welcomed by patients and other stakeholders. What is often overlooked however, is the impact FDA approvals have on patients and drug development outside of the US.
The FDA’s gatekeeping power
The primary role of the FDA is to protect public health by ensuring the benefits of new medical products outweigh their risks. The FDA also provides information to patients and prescribers and strengthens incentives for firms to develop drugs to treat a variety of medical conditions. The gatekeeping power of the FDA to ultimately decide how manufacturers develop their products, which new products enter the market, and which firms are successful is unparalleled globally. It is therefore unsurprising that the FDA is referred to by some as the “most powerful regulator in the world”.
Today, the FDA oversees the largest single market for pharmaceuticals globally, worth an estimated $528 billion in 2020 in public and private spending (Figure 1). Though the reach and influence of the FDA extends far beyond just the US market.
Figure 1 – Estimated net pharmaceutical manufacturer revenue by country, 2020

Source: IQVIA, 2021: Global Medicine Spending and Usage Trends: Outlook to 2025
Why is the FDA so influential globally?
In most cases, pharmaceutical research and development occurs once globally. Clinical trials used to demonstrate a single product is safe and efficacious are conducted once, with data from these trials submitted to different regulators globally for product approval in different markets. Being the largest market, that also pays the highest prices, the US market is by-far the most lucrative for manufacturers. As a result, evidence development for new drugs is almost always generated primarily to meet FDA approval standards and submitted to the FDA for approval before being submitted to other regulators globally. Effectively, the FDA sets the bar in terms of entry standards for drug approvals and for incentives for drug development globally (and not just in the US).
Fortunately, the FDA is generally a highly capable regulator that has a reputation for scientific excellence and the most rigorous approval standards, though this has not always been the case. Prior to 1938, there was no legal requirement globally for studies to be conducted on new medicines. Until 1937, sulfanilamide had been widely and successfully used in powder and tablet form to treat streptococcal infections. When adapted to a more-convenient liquid form, 70 percent diethylene glycol was used (a toxic chemical used in antifreeze and brake fluid). The resulting deaths of over 100 people in the US ultimately led to the 1938 Federal Food, Drug, and Cosmetic Act (FD&C Act) which required manufacturers to demonstrate drug safety in clinical trials. Provisions under the FD&C Act later protected American citizens (other than those who had participated in clinical testing) from the Thalidomide disaster in which an estimated 5-10,000 babies were born with severe deformities around the world outside of the US in the late 1950s and early 1960s. These, and other events, effectively strengthened the FDA mandate to regulate new drugs coming onto the market.

FDA Campus Exterior Shot by The U.S. Food and Drug Administration is United States government work
Since then, approval standards set by the FDA have become the model on which pharmaceuticals are regulated globally, with approval decisions by the FDA predictably having high concordance with other regulators’ globally.
Why is FDA global influence so important?
But the FDA is not perfect. Recently, some high-profile approval decisions have led to speculation whether the FDA has, at times, placed the special interests of industry ahead of patients’. It is not only experts that are concerned, polls also show that public trust in the FDA is waning.
Looking at recent FDA drug approvals, there are trends which, given the global influence of the FDA, other regulators should also take heed of. As Figure 2 shows, the 5-year average number of annual FDA approvals currently sits at 51 drugs per year (over double compared to a decade ago). More drugs on the market seemingly leads to better treatment options for patients – but not necessarily. Increasingly, we see a greater number of treatment options in crowded therapeutic areas with little or no added benefits which are already difficult to discern in the first instance. This complicates reimbursement decisions for payers in other countries that are tasked with the increasingly difficult choice of which new drugs to fund, and at what cost under growing budgetary constraints.
Figure 2 – FDA drug approvals per year, 2000-2021
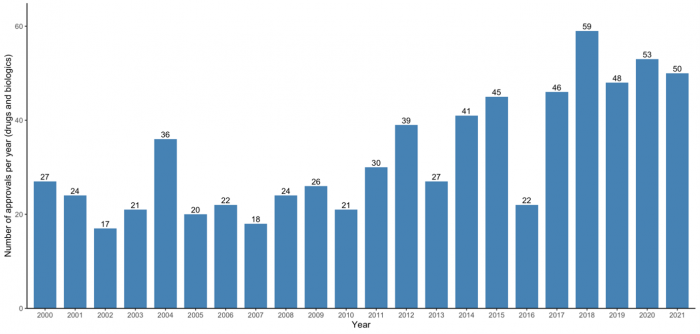
Source: Adapted from: Mullard (2022): 2021 FDA Approvals
Though cancer represents a major global burden of disease, it does not represent the largest disease burden globally, particularly in low-and middle-income countries. Despite this, cancer drugs dominate drug approvals in the US accounting for 30 percent of them in 2021. Some of the mismatch between drugs developed and the unmet global health need may partly be attributed to market dynamics in the US where there is a higher willingness-to-pay for cancer drugs, along with federal policies which prevent some public insurers from negotiating cancer drug prices. These, and other factors, produce a large pull-through effect for cancer drug development compared to other disease areas.
In recent years, drugs are being approved more quickly, with greater uncertainty of their benefits and risks. On the one hand, this can potentially benefit some patients by providing access to treatments. On the other it can reduce the quality and quantity of evidence on their benefits and risks, leading to greater uncertainty for payers and other future patients. It is the task of regulators, like the FDA, to find the right balance between patient access and uncertainty. Ethically and scientifically, this is no easy task for the FDA with some evidence suggesting the correct balance is not always found. Once approved by the FDA, other regulators globally are placed under significant political pressure to approve these drugs in their respective regions.
The FDA is much more influential than other drug regulatory agencies
Actions of the FDA have vast knock-on effects for drug development and approvals for patients and the public not just in the US but globally. Mechanisms of global drug development, market dynamics and other policy issues result in the FDA largely deciding which new drugs are developed by firms, how they are developed and the quality and quantity of evidence available outside of the US.
- Please read our comments policy before commenting.
- Note: This article gives the views of the author, and not the position of USAPP – American Politics and Policy, nor the London School of Economics.
- Shortened URL for this post: https://bit.ly/3GVeIme